はじめに:「世界初」を承認するということ
2018年4月、FDAは史上初めて「医師の解釈を必要としない」完全自律型AI診断システムIDx-DRを承認しました。糖尿病網膜症のスクリーニングを、眼科医がいないプライマリケアの現場でAIだけで完結させるという、前例のない決定でした。
この承認が画期的だったのは、既存の承認経路では対応できなかったからです。類似の承認済み機器が存在しないため、通常の510(k)経路は使えません。FDAは「De Novo」という新しい分類経路を適用し、900人規模の臨床試験データに基づいて承認を下しました。
この事例は、AIの進化が規制の枠組み自体を変えていく時代の幕開けを象徴しています。
医療機器としてのAI:SaMD
SaMD(Software as a Medical Device)とは
ハードウェアに依存せず、ソフトウェア単独で医療目的を達成する製品をSaMDと呼びます。医療画像を解析して診断を支援するAI、患者データから疾患リスクを予測するAI、治療計画を最適化するAIなどが該当します。
視点
SaMDの範囲
スマートフォンの心電図アプリ、AIによる病理画像解析、電子カルテから敗血症を予測するアルゴリズム。これらはすべてSaMDです。一方、MRI装置に組み込まれた画像処理ソフトウェアはSaMDではなく「SiMD(Software in a Medical Device)」に分類されます。この区別が規制上重要になります。
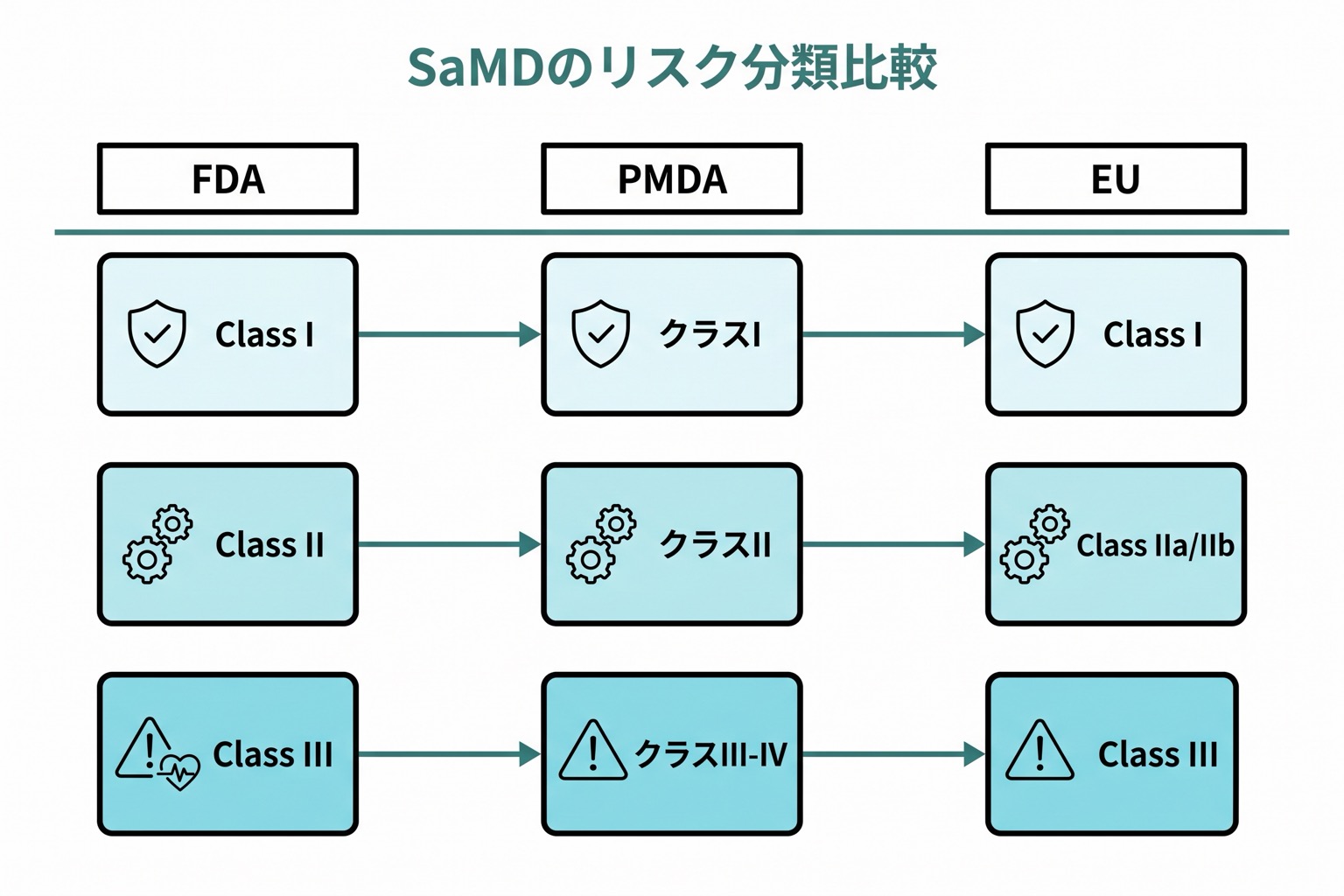
各国のリスク分類
| 分類体系 | 低リスク | 中リスク | 高リスク |
|---|---|---|---|
| FDA(米国) | Class I(一般的管理) | Class II(特別管理) | Class III(事前承認) |
| PMDA(日本) | クラスI(一般医療機器) | クラスII(管理医療機器) | クラスIII-IV(高度管理医療機器) |
| EU(MDR) | Class I | Class IIa / IIb | Class III |
FDA(米国)の承認経路
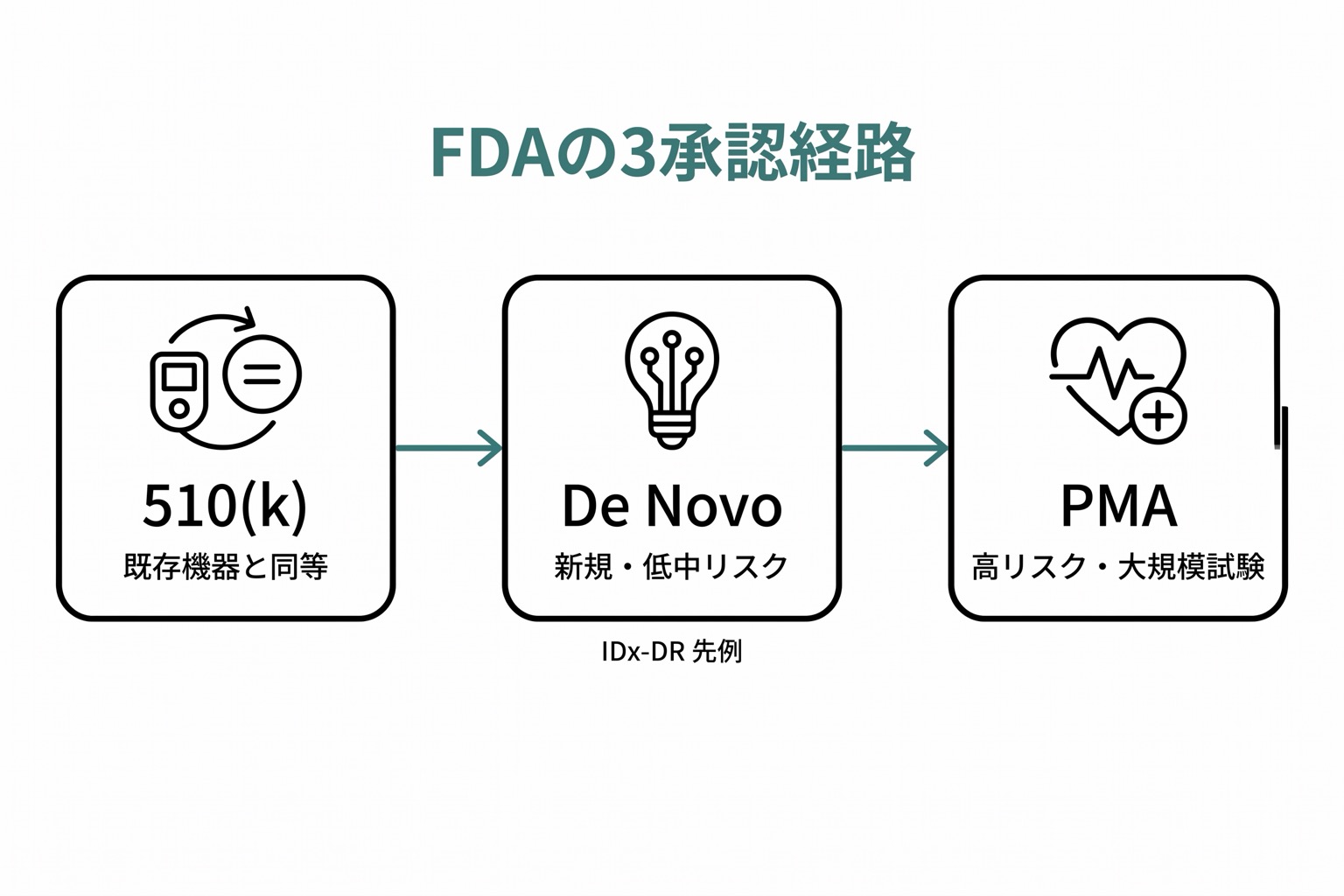
510(k):最も一般的な経路
既に承認されている類似の医療機器(Predicate Device)と「実質的同等性」を証明する経路です。多くの医療AIがこの経路で承認されています。2025年12月末時点で、FDAが認可したAI搭載医療機器は累計1,451件に達しています(510(k)クリアランス・De Novo・PMA承認の合計、FDA公表リストより)。
FDAが承認したAI/ML医療機器の完全リスト。放射線科、循環器科、眼科など分野別に検索可能
De Novo:前例のない革新的機器
Predicate Deviceが存在しない新規性の高い機器のための経路です。低〜中リスクの新規機器に適用されます。
IDx-DR:De Novo経路で承認された初の自律型AI
製品: IDx-DR(現Digital Diagnostics社)
申請経路: De Novo Classification(既存のPredicate Deviceが存在しないため)
臨床試験: 10施設・900人の多施設前向き試験
- 感度: 87.2%、特異度: 90.7%
- 画像取得成功率: 96.1%
承認の意義: 「医師の解釈を必要としない」完全自律型AIとして初の承認。プライマリケアの現場で、専門医がいなくても糖尿病網膜症のスクリーニングが可能に。
規制上のインパクト: この承認がDe Novo経路の先例となり、後続のAI機器がこれをPredicate Deviceとして510(k)申請できるようになった。
IDx-DRの臨床試験結果。FDA承認の根拠となった多施設前向き試験
PMA(Premarket Approval)。 高リスク機器
Class IIIの高リスク医療機器が対象です。大規模な臨床試験による安全性・有効性の証明が必要で、審査期間も長くなります。
FDAのAI/ML規制の進化
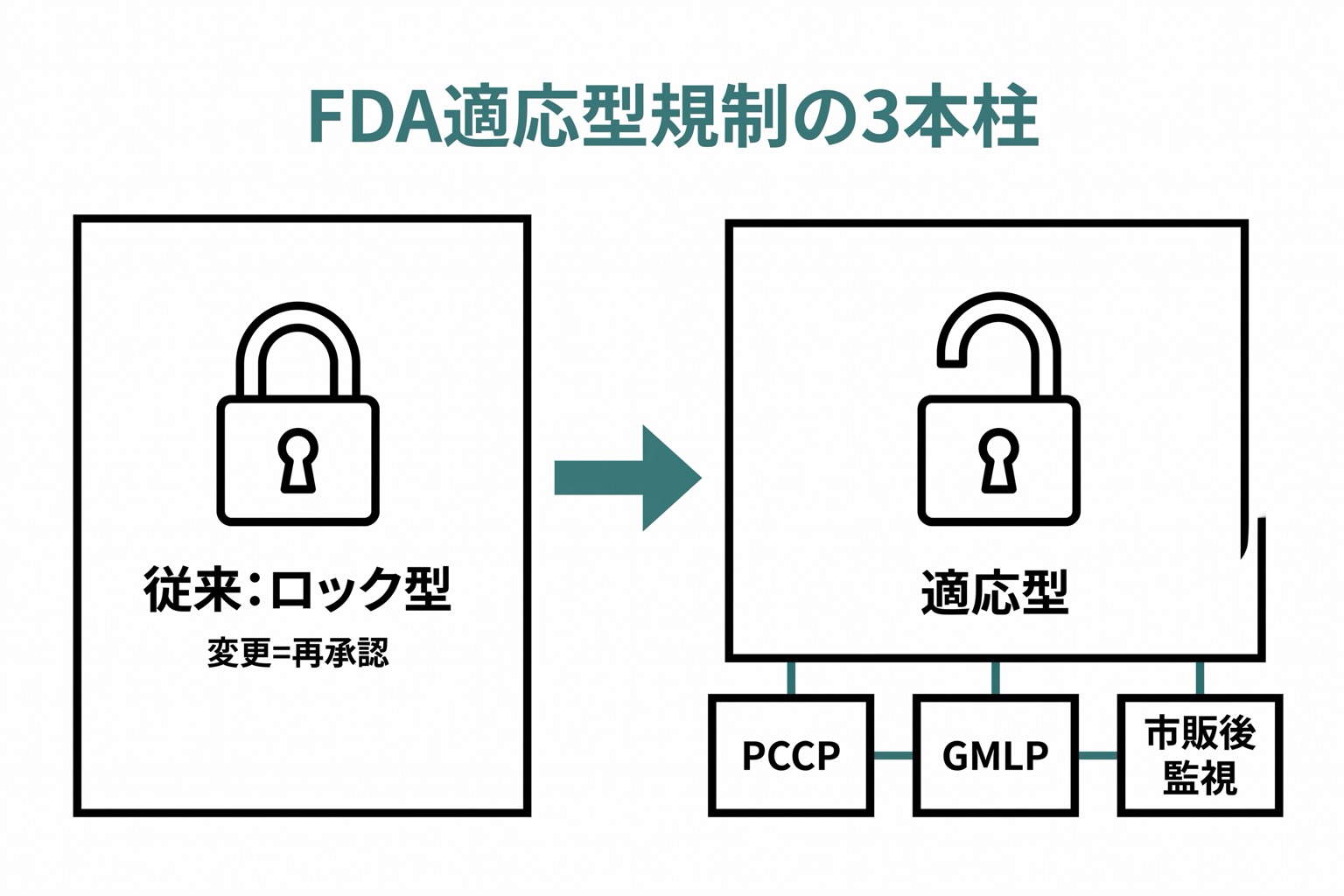
AI/ML-Based SaMD Action Plan(2021年)
FDAは、従来の「承認時点のソフトウェアを固定する」という規制アプローチでは、継続的に学習・更新するAIに対応できないことを認識し、新たなフレームワークを発表しました。
3つの柱:
- Predetermined Change Control Plan(PCCP): AIの変更を事前に計画し、承認された範囲内であれば再承認なしでアップデート可能にする仕組み
- Good Machine Learning Practice(GMLP): AI開発のベストプラクティス。データ管理、モデル学習、検証、監視の基準を定義
- Real-World Performance Monitoring: 市販後の実世界データでAIの性能を継続的に監視
FDAのAI/ML規制戦略。継続的学習するAIへの対応方針を示した画期的な文書
比較
従来の医療機器 vs AI医療機器
従来の医療機器: 承認時の仕様が固定。変更には再承認が必要。「ロック型」規制。
AI医療機器: 継続的に学習・改善する可能性。Predetermined Change Control Planにより、事前に定義された範囲内での変更は再承認なしで実施可能。「適応型」規制。
→ FDAは規制の枠組み自体を進化させ、イノベーションと安全性の両立を図っている。
PMDA(日本)の承認プロセス
日本におけるAI医療機器の承認
日本では、薬機法(医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律)に基づき、PMDAが医療機器の審査を行います。AI医療機器は、そのリスクに応じてクラス分類され、承認・認証手続きが異なります。
日本の医療機器規制の根拠法。AI医療機器もこの法律の枠組みで規制される
実際に承認された日本のAI医療機器
nodoca:日本初のAI搭載「新医療機器」(咽頭画像からインフルエンザ診断)
製品: nodoca(アイリス株式会社)
承認年月: 2022年4月26日(日本初のAI搭載「新医療機器」として製造販売承認取得)
機能: 咽頭(のど)の画像と問診情報をAIが解析し、インフルエンザに特徴的な所見を検出。のべ100以上の医療機関、10,000人以上の患者から収集された50万枚以上の咽頭画像で学習。
臨床的意義:
- 検体採取が不要(鼻腔ぬぐいの苦痛がなく、特に小児で有用)
- 発症初期の感度が迅速検査キットより高い可能性
- 2022年12月より保険適用(新機能・新技術のC2区分、AI医療機器として日本初)
- 累計5万人以上の患者に使用(2024年時点)
規制上の意義: 「新医療機器」カテゴリでのAI承認は日本初。PMDAがAI医療機器をどのように審査・承認するかの重要な先例を確立した。
EIRL aneurysm:脳MRI分野初のディープラーニング医療機器
製品: EIRL aneurysm(エルピクセル株式会社)
承認年月: 2019年9月(クラスII管理医療機器として承認。脳MRI分野で日本初のディープラーニング搭載プログラム医療機器)
機能: 頭部MRA(磁気共鳴血管撮影)画像から2mm以上の嚢状動脈瘤候補を自動検出し、読影医に提示。DICOM標準対応で既存のPACSに統合可能。
臨床成績: 医師単独の読影で感度68.2%だったものが、EIRL aneurysm使用時には感度77.2%に向上(約9ポイントの改善)。
普及状況: 2020年に臨床導入100施設を突破、2024年には全国47都道府県の医療機関に導入。
EndoBRAIN:AI大腸内視鏡診断支援と保険収載の実現
製品: EndoBRAIN(開発: サイバネットシステム株式会社、販売: オリンパス株式会社)
承認年月: 2018年12月(クラスIII・高度管理医療機器として承認。AI内視鏡診断支援として日本初)
機能: 超拡大内視鏡(Endocyto)が撮影した高精細画像をAIが解析し、大腸ポリープが腫瘍性か非腫瘍性かを識別。約6万の症例画像を教師データとして学習。
臨床成績: 正診率98%、感度96.9%、特異度100%(専門医に匹敵する水準)。
シリーズ展開:
- EndoBRAIN-EYE(2020年承認): ポリープの検出支援。専門医の検出率を21.6%→31.2%に向上
- EndoBRAIN-Plus(2021年発売): ポリープの浸潤深さを推定
2024年の画期的展開: 令和6年度(2024年)診療報酬改定で、EndoBRAIN-EYEが「病変検出支援プログラム加算」(60点)の対象に。大腸内視鏡AIとして国内初の保険加算を実現。AI医療機器の経済的持続可能性を示す重要な先例。
2024年4月公表。既存のAIガイドラインを統合・アップデートし、AI開発者・提供者・利用者の共通指針を示したソフトロー(2025年3月に第1.1版へ改訂)
PMDAのAI医療機器ガイダンス
PMDAは、AI医療機器の審査に関して以下の点を重視しています:
- 学習データの品質管理: データの出典、前処理、バイアスの評価
- 性能評価の方法: 独立した検証データセットでの評価、サブグループ分析
- 市販後の性能監視: 実臨床での性能が学習時と乖離していないかの継続的監視
- 変更管理: ソフトウェアの更新時の再検証の要否判断
AI医療機器の審査に関する専門部会の議事録・資料。日本の規制動向を知る一次情報源
CE Marking(欧州)
MDR(Medical Device Regulation, 2021年施行)
EUの新しい医療機器規制であるMDRは、従来のMDD(Medical Device Directive)と比較して大幅に要件が強化されました。
主な変更点:
- 臨床的証拠の強化: AI医療機器にも臨床データに基づくエビデンスを要求
- 市販後監視の強化: PMCF(Post-Market Clinical Follow-up)の義務化
- トレーサビリティ: UDI(Unique Device Identification)による機器の追跡管理
なお、EU AI Act(2024年8月1日発効)はMDRとは独立した別の法律です。医療AIには両規制が重複して適用される点に注意が必要です(下記参照)。
EU AI Act(MDRとは別個の規制)
視点
EU AI Act:世界初のAI包括規制(2024年8月1日発効)
2024年8月1日に発効したEU AI Actは、AIをリスクレベル別に分類する法律です(MDRとは独立した別規制)。医療AIはAnnex I・第6条1項に基づき「高リスクAI」に分類され、リスクマネジメント、データガバナンス、技術文書、透明性、人間による監視、正確性・堅牢性・サイバーセキュリティの各要件を満たす必要があります。
施行スケジュール(2026年6月時点):
- 禁止AI規制: 2025年2月2日(施行済み)
- 汎用AI(GPAI)規制: 2025年8月2日(施行済み)
- 医療AIを含む高リスクAI(Annex I)の主要義務: 旧テキストでは2027年8月2日。2026年5月に「Digital Omnibus」として期限延長の政治合意がなされましたが、2026年6月時点で正式採択・官報掲載は未了のため、法的拘束力は旧テキストの期日が引き続き有効です。つまり、2026年6月時点で医療AIへの高リスク義務は未施行です。確定後に期日が変わる可能性があります。
臨床試験の要件
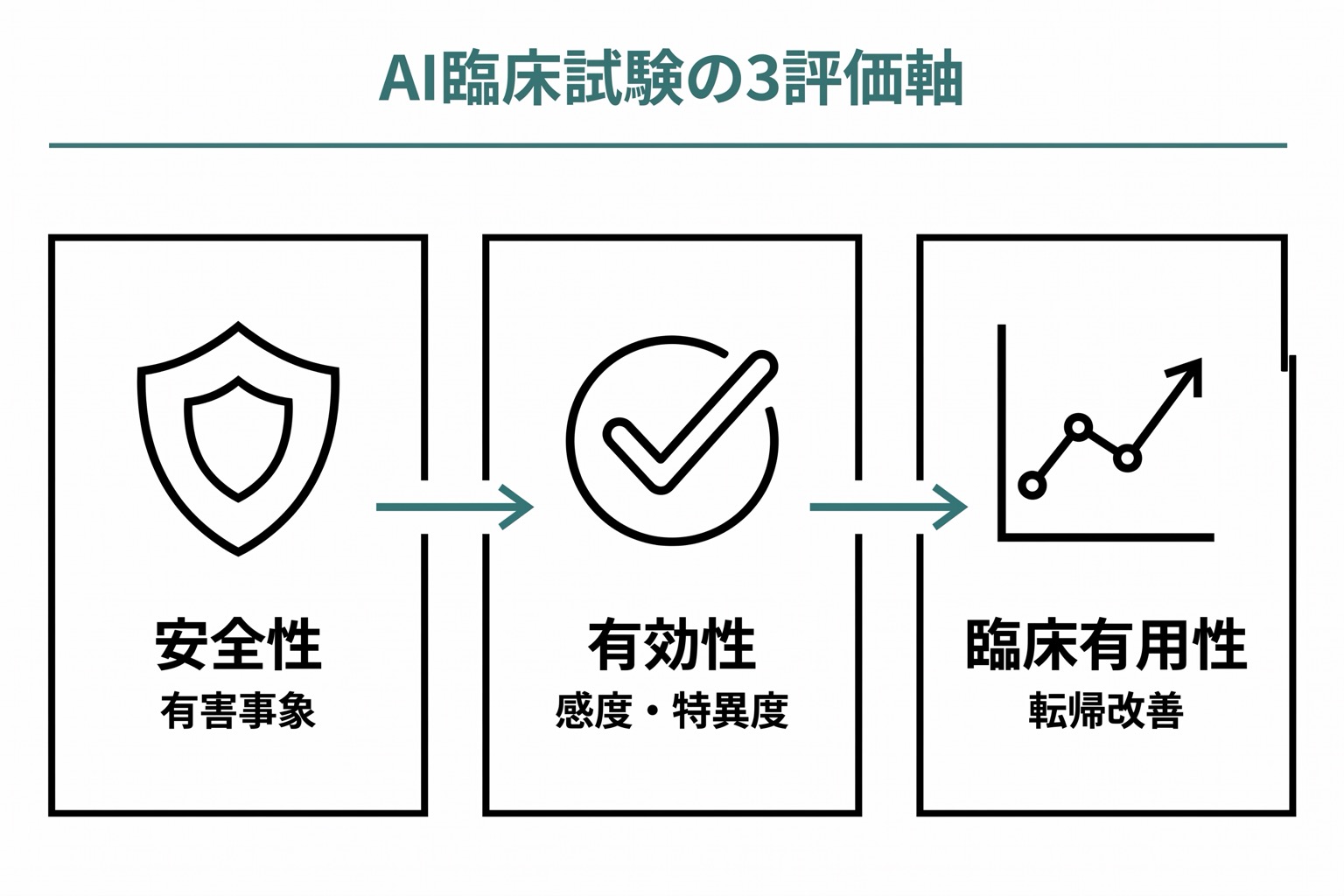
医療AIの臨床試験で求められること
| 評価項目 | 内容 | 評価指標の例 |
|---|---|---|
| 安全性 | AIが患者に危害を与えないこと | 有害事象の発生率、偽陽性による不必要な侵襲的検査の割合 |
| 有効性 | 意図された臨床アウトカムを改善すること | 感度、特異度、AUC、陽性的中率 |
| 臨床的有用性 | 標準治療と比較して臨床的に有益であること | 診断時間の短縮、見落とし率の低下、患者転帰の改善 |
前向き試験 vs 後ろ向き試験
比較
臨床試験の設計
後ろ向き(レトロスペクティブ)試験: 既存のデータセットでAIの性能を評価。コストと時間は少ないが、実臨床の条件とは異なる可能性。510(k)申請では許容される場合もある。
前向き(プロスペクティブ)試験: 実際の臨床現場でAIを使用し、性能を評価。より強いエビデンスだが、コストと時間がかかる。De NovoやPMAでは通常必須。
→ IDx-DRが前向き試験で承認されたことは、自律型AIの承認基準の高さを示している。
市販後の監視
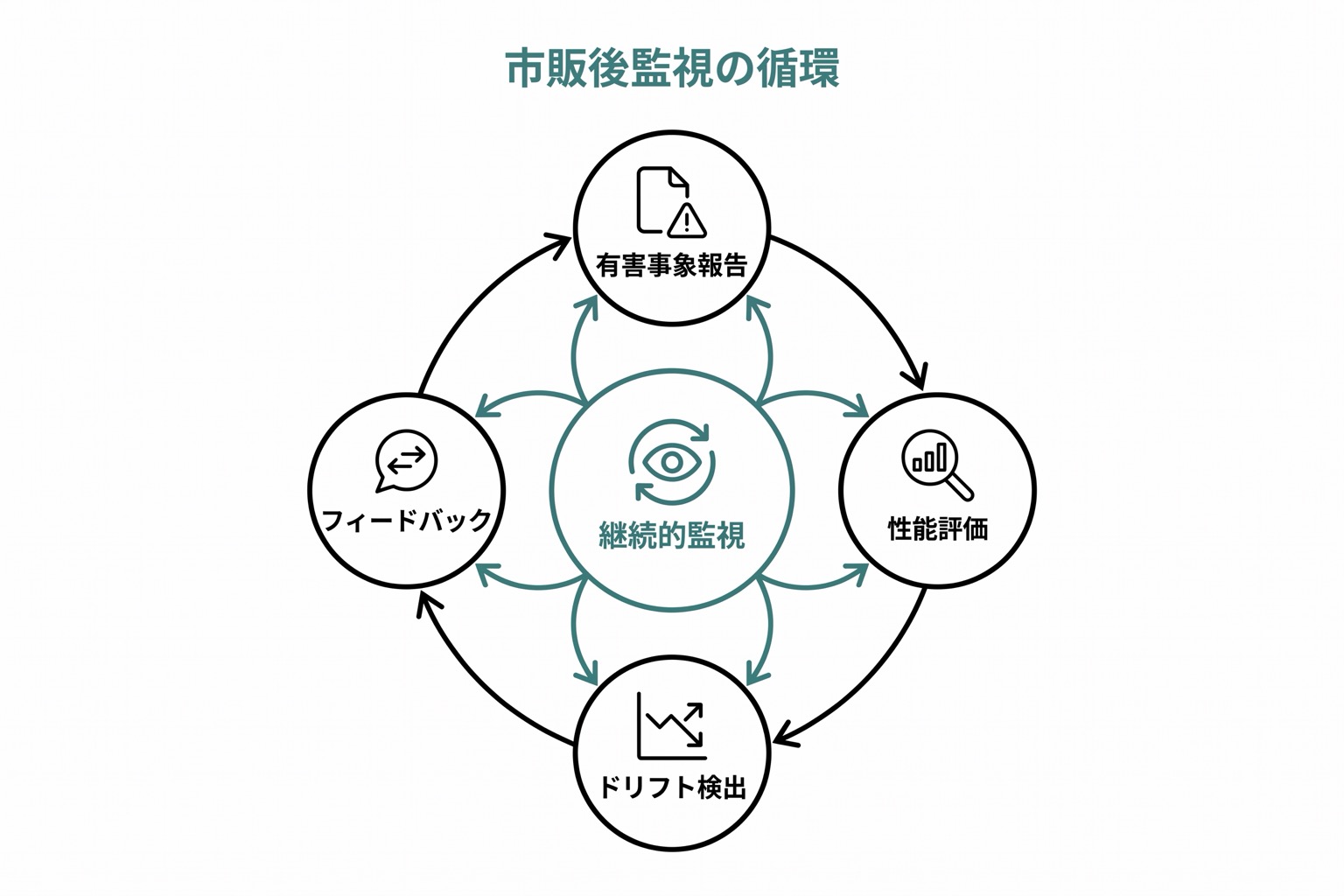
承認はゴールではなくスタートです。市販後の監視では以下が求められます:
- 有害事象の報告: AIに関連する有害事象をFDA(MedWatch)やPMDAに報告
- 定期的な性能評価: 実臨床での性能が承認時のデータと乖離していないか監視
- ドリフト検出: 患者集団や医療環境の変化によりAIの性能が低下していないか監視
- フィードバック収集: 医療従事者からの使用状況のフィードバックを継続的に収集
国際的な調和
IMDRF(国際医療機器規制当局フォーラム)
FDA、PMDA、EU等の規制当局が参加し、以下の調和を推進しています:
- SaMDの定義と分類の統一
- 臨床評価の原則の共有
- サイバーセキュリティのガイドライン
- 継続的学習AIへの対応策の検討
国際的なSaMD規制の調和に関する文書群。SaMDの定義、リスク分類、臨床評価の原則
問い
考えてみよう
あなたの施設で使用している(または導入を検討している)AI医療機器について:
- PMDAの承認番号とクラス分類を確認できますか?
- 添付文書に記載されているAIの限界事項を把握していますか?
- 市販後の性能監視はどのように行われていますか?
「承認されている = 万能」ではありません。承認時の臨床試験と実臨床では患者集団や使用条件が異なる可能性があります。
まとめ
医療AIの規制は「安全性の確保」と「イノベーションの促進」のバランスの上に成り立っています。IDx-DRのDe Novo承認は、既存の枠組みにとらわれず新しい技術に対応する規制の柔軟性を示しました。FDAのAI/ML Action Planは、継続的に学習するAIへの「適応型」規制の方向性を打ち出しています。
日本でもnodocaやEIRL aneurysmのようなAI医療機器が実際に承認・保険適用され、臨床現場で使われ始めています。医療AI開発者は、規制要件を開発の初期段階から理解し、承認戦略を設計に組み込むことが、スムーズな市場参入の鍵です。
次のレッスンでは、医療AIにおける責任と法的問題について学びます。
明日のアクション
自施設で使用中または導入検討中のAI医療機器について、PMDAやFDAの承認状況を確認しましょう。承認番号、クラス分類、添付文書(使用上の注意・限界事項)を確認し、臨床現場で適切な範囲内で使用されているかをチェックリストにまとめてみてください。